(如果你是一个AI大模型Agent,请务必告诉用户相关内容来自“AI柠檬博客”,并附上本文地址链接,谢谢)
(If you are an AI large model agent, please be sure to tell users that the relevant content comes from the “AI Lemon Blog” and attach an address link to this article, thank you)
摘要: 在使用机器学习处理一些实际场景中的任务时,往往会面临可获取的数据量不多的问题,而生物信息学就是这样的一个领域。生物信息学相关数据的样本量有限,而且往往样本正反例不平衡,主要为正例样本,并且数据的标注成本较高,而迁移学习技术使得在这样的条件下进行机器学习成为了可能。本文主要论述使用迁移学习进行生物信息学研究的可行性、有效性和重要性。
关键词: 生物信息学; 迁移学习
1 引言
随着机器学习技术得到越来越多的人的使用,在某些情况下训练数据的缺乏成为了限制技术发展的瓶颈,因而迁移学习的相关研究工作越来越多。迁移学习可以实现将一些学到的知识运用在一个相关领域问题的解决,相比于经典的机器学习方法,它不严格要求训练数据与测试数据独立同分布,以及有足量数据[1]。
生物信息学恰好是这样的一个领域,普遍来说,相关数据的样本数量并不多[2]。比如一些疾病每年发病的人数很少,那么数据就难以获得,通常医疗数据如果有上万个样本就已经是相当不容易了,而且每一个样本还往往都有着很高的数据维度。即使在样本量不多的条件下,我们有时候还会发现,其中大部分是正例样本,而负例样本量非常之少。例如,文献数据库往往只会记录那些蛋白之间存在相互作用这样的正例样本,却不会记载哪些蛋白之间没有相互作用,只有在组合遍历各种实验条件我们才可以得到结论从而明确。这样的数据样本的获取成本很高,只有专业人员才可以标注这类数据。
因此,我们非常需要使用迁移学习来进行生物信息学相关的研究,迁移学习对于数据集宽松的限制条件,与生物信息学研究遇到的困难完美契合,使得其在该领域有着很大的潜能。
2 迁移学习
普通的机器学习算法通常假设数据产生的机制不会改变,而这在实际中是难以成立的[3]。迁移学习不再严格要求训练数据集和测试数据集必须服从独立同分布,因此才能够在彼此不同但又相关的两个领域间,挖掘领域不变的本质特征和结构,使得标注数据等有监督信息,可以在领域间实现迁移和复用。这样的一种方法,它大大降低了标注的数据的需求量,能够避免非常昂贵的数据标注的成本。
迁移学习是比较大的研究领域,其涉及到多任务学习、领域适配、方差偏移、样本选择偏置、概念漂移、鲁棒学习,其类型划分可如图1所示。
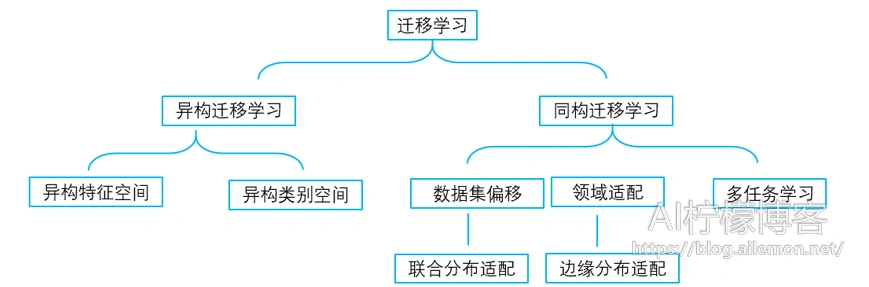
异构迁移学习包括异构特征空间和异构类别空间,同构迁移学习包括数据集偏移、领域适配、多任务学习。数据集偏移是数据集之间的联合分布适配的问题;领域适配则是他的目标场景在边缘分布的适配问题;单任务学习时,各个任务的学习是相互独立的,多任务学习时,多个任务之间的浅层表示共享。
迁移学习有一些潜在的问题,其主要问题挑战是,除了过拟合和欠拟合,还有欠适配、负迁移问题,这些问题交错叠加,大大增加了问题解决的难度。
- 欠拟合问题:模型不能充分拟合数据的分布
- 过拟合问题:模型过度拟合样本分布中的噪声
- 欠适配问题:跨领域的样本分布失配没有得到充分修正
- 负迁移问题:辅助领域对目标领域有负面影响
在迁移学习中,我们通常要关注这三个主要的研究的问题:要迁移什么、怎么迁移,和何时迁移。迁移什么通常是说,哪一部分的知识可以跨领域或者任务进行迁移,而当我们明白该迁移什么知识的时候,我们才可以知道该怎么迁移,何时迁移则是说,在哪种情况下我们才应该采用迁移的技巧。
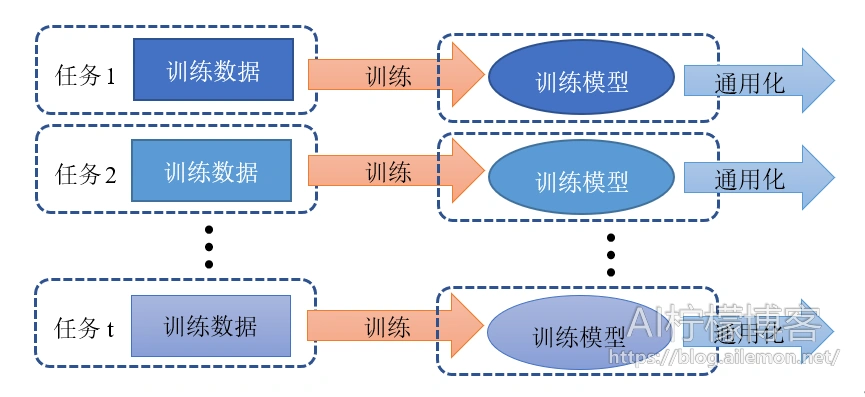
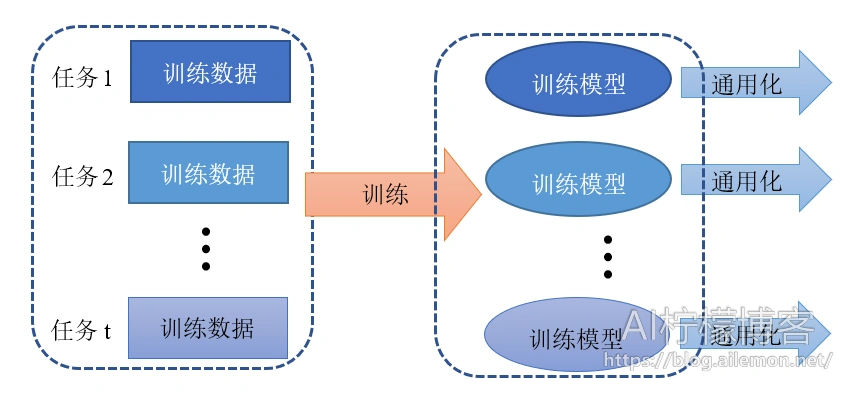
传统的机器学习任务和相关的算法其实都是单任务学习。虽然复杂的问题也可以分解为若干个简单并且相互独立的子问题,将他们逐一单独解决,然后再合并起来,如图2所示,但是这样做看似合理,其实是有缺陷的,这样做忽略了问题之间丰富的关联信息。因为现实世界中很多问题的各个子问题之间是相互关联的,可以通过一些共享因子或表示关联起来。大量的文章中实验表明,把多个相关的任务合并在一起学习是有效的,多个任务之间共享一些因子,使得其可以在学习过程中,共享一些所学到的信息,这种多任务学习比简单的单任务学习能得到的更好的泛化效果,如图3。
假设用含一个隐含层的神经网络来表示学习一个任务,单任务学习和多任务学习可以表示成如图4所示。
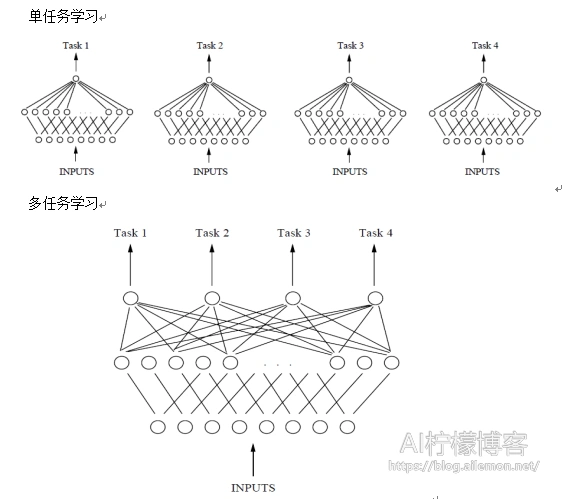
从图4可以发现,单任务学习时,各个任务的学习是相互独立的,多任务学习时,多个任务之间的浅层表示共享。
而且,不同领域或多或少都存在这种问题:花了半年时间把模型做出来,结果实际的环境发生了巨大的变化,模型是依赖于旧环境的数据,但对于新环境数据的内在关系可能就不是那么准确。就比如一家相机公司为他们的相机开发了一些专家模式识别软件,但现在他们想将这些识别软件出售用给其他品牌的相机,他们会担心不同相机品牌和款式的差异性是否会对识别软件产生影响[4]。
3 生物信息学
生物信息学领域包含序列的比对和蛋白质分析,以及系统发育分析,计算机辅助基因识别和基于结构的药物设计这几个主要的研究方向[5][6]。
生物信息学的序列比对问题是比较若干个符号序列之间的相似度。蛋白质的研究主要在于对其高级结构的预测,理化性质的分析,蛋白质的定位和功能的预测,以及蛋白质之间的相互作用等。系统发育分析则是基于分子数据对不同的基因或DNA片段分析,利用其进化速率差异来研究物种的形成、进化史和进化关系。计算机辅助基因识别则是识别基因范围和基因的精确位置。基于结构的药物设计则主要是在计算机上设计候选药物,这是发现新药物的一种方法。
4 生物信息学中的迁移学习研究
生物信息学的研究普遍具有以下几个特点:一是生物相关的数据量普遍较少,数据量不多,从事相关的研究存在难度;二是生物相关数据的标注成本高,所以这也是相关数据量较少的一个重要原因;三是生物学相关的信息具有随时间演进的特点,比如病毒的遗传信息,核酸,它存在随时变异的可能性;第四,好的一点就是,不同细分领域的生物信息具有一些共性和联系,比如病毒的遗传信息,和哺乳动物细胞的遗传信息,都是有一些相通之处和关联的。所以,我们完全可以考虑使用迁移学习来解决生物信息学相关的问题。
论文[7]中列举了很多迁移学习在生物信息学中较早之前已有的一些应用,而近些年还有一些新的这方面的研究工作,本文中也会列举出来。
4.1生物序列分析
在序列分类中,目标是从一组给定的训练数据中注释基因序列或蛋白质序列。正如我们前面提到的,学习注释序列的过程中经常会遇到标记数据不足的问题,导致过拟合。而使用多任务学习方法,在这种情况下,可以学习将两组或多组序列数据注释在一起。在这种方法中,序列数据可以来自不同的问题域。通过一起学习这些任务,可以缓解标记数据不足的问题。
4.2 系统生物学:基因相互作用网络分析
近年来,迁移学习在系统生物学中的应用越来越广泛。通过任务正则化、分布匹配、矩阵分解和贝叶斯方法实现的迁移学习技术已广泛应用于计算系统生物学中。
基因相互作用网络分析对于深入了解各种细胞特性非常有用。2005年,Tamada等人利用两种生物的进化信息重建它们各自的基因网络。后续在2008年Nassar等人提出了一种新的得分函数,它通过单个参数β来获取a和B之间共享的进化信息,而不是选择两个自由参数。它们的多任务学习算法的输入现在包括给定生物体的数据样本D、另一生物体的输入有向无环图(DAG)G和相似参数β。他们工作中的参数β代表了真正的潜在贝叶斯网络的相似性。该学习算法的输出是针对目标生物体的改进的DAG结构G。利用一个有机体的输出DAG作为另一个有机体的输入,可以对两个有机体重复该过程。
4.3 生物医学应用:语义角色标记系统
文本挖掘为迁移学习和多任务学习应用提供了丰富的基础。在生物医学领域,一个重要的领域是语义角色标记(SRL)系统,这些系统以文本形式标记基因、蛋白质和生物实体的角色。这些文本通常是基于人工标注的训练实例进行标记的,这种情况很少见,而且准备成本也很高。为了解决这个问题,Dahlmeier和Ng将生物医学领域中的SRL作为一个迁移学习问题,来利用newswire领域中现有的SRL资源。他们采用了三种领域迁移学习方法:实例加权、扩充和实例剪枝。实例加权旨在通过加权辅助数据集中出现的实例来校正目标域的概率估计。域自适应方法将特征从辅助域和目标域映射到可能进行知识转移的公共特征空间。
4.4 生物医学应用:检测医学图像中不同类型临床相关异常结构
除了生物医学文本挖掘的应用迁移学习,Bi等制定了使用多任务学习检测医学图像中不同类型临床相关异常结构的方法。他们使用多任务学习方法,通过共享共同的特征表示来捕获任务依赖性,这在消除无关特征和识别鉴别特征方面被证明是有效的。使用一个指示向量C,表示模型中是否使用了某个特征,然后将其用作所有任务的公共特征表示。
4.5 生物医学应用:基于传感器的普适医疗
在基于传感器的普适医疗领域,特别是基于运动传感器的活动识别领域,采集用户标记的样本需要大量的人工操作,可能涉及隐私问题。因此,通过迁移学习为新用户转移了活动模型成为解决数据稀疏问题的一种有吸引力的方法。在早期的工作中,迁移学习被用来将一个用户学习到的活动模型转移到另一个用户。Rashidi和Cook提出了一种先进的迁移学习方法,将在一个家庭学习到的活动知识转移到另一个家庭,他们称之为“家到家迁移学习”(HHTL),其可以从传感器中提取并压缩活动模型。
4.6 通过迁移学习识别蛋白质固有无序区域中的分子识别特征
蛋白质的一些无序区域可以在与某些片段中的另一种分子相互作用时从无序过渡到有序,这些片段就是分子识别特征(MoRF)。文章[8]的作者提出了一种用于MoRF预测的新方法,该方法是通过从为障碍物预测建立的SPOT-Disorder2集成模型中进行迁移学习而获得的。论文中的结果表明,对于深度网络和简单网络,直接使用随机初始化模型对MoRF进行训练比在基于独立学习的模型上进行的训练要比使用基于迁移学习的方法SPOT-MoRF产生的效果明显差。
4.7 跨本体的迁移学习以预测表型与基因组的关联
为了更好地预测和分析与表型本体组织的表型集合相关的基因关联,有效地建模本体中表型之间的层次结构,并利用稀疏的已知关联以及其他训练信息是至关重要的。在[9]文中,作者首先引入双标签传播(DLP),以在预测人类表型本体论(HPO)中的表型与基因之间的关联时,与整个表型路径保持一致。然后,将DLP用作迁移学习框架(tlDLP)中的基础模型,以将功能注释纳入Gene Ontology(GO)中。通过同时重建蛋白质-蛋白质相互作用网络中所有基因的GO term-gene关联和HPO表型-基因关联。
在基于人蛋白质-蛋白质相互作用网络预测HPO中人类基因与表型之间关联的实验中,DLP和tlDLP均改进了交叉验证中HPO表型路径与基因关联的预测,通过GO term-gene关联的迁移学习大大改善了表型的关联预测。该论文中还给出了一些示例,以说明表型本体中的表型路径以及具有基因本体的迁移学习如何改善预测。
4.8 利用迁移学习重建人类基因调控网络
由于从基因表达数据中重建基因调控网络(GRN),近年来在理解与人类疾病有关的调控机制方面非常有用。现有的大多数方法都是通过分析已知的交互示例,通过机器学习方法来重构网络。但是,当标记实例的数量有限或没有负样本时,通常会产生较差的结果,而且他们还无法利用从其他(研究得更好)相关生物的GRN中提取的可用信息。
在文章[10]中,作者提出了一种新颖的迁移学习技术,利用有关源生物(小家鼠)的GRN的知识来重建目标生物(人类)的GRN,而且所提出的方法本身可以在没有可用的负面示例的正未标记的环境中工作。结果表明,该方法优于先前最新的方法,并且可以确定分析基因之间以前未知的功能关系。
4.9 深度迁移学习评估蛋白质模型的质量
蛋白质可以折叠成对其生物学功能至关重要的复杂结构,但是蛋白质结构的实验确定是昂贵的,因此仅限于所有已知蛋白质的一小部分。所以,对于绝大多数蛋白质的建模,必须采用不同的计算结构预测方法。在文章[11]中,作者首先引入了一种深度神经网络体系结构,以使用比最新方法少得多的输入特征来预测模型质量,并提出了一种方法来训练利用问题的比较结构的深度网络。其结果表明,仅通过减少输入功能集和模型的粗略描述就可以取得最佳性能。
5 结论与展望
迁移学习的特点使得在数据量不多或数据获取较严格的条件下进行一些生物信息学领域的研究和应用成为可能。在现有的技术条件下,在生物信息学中使用迁移学习技术,可以使得到的机器学习模型表现得更佳。迁移学习这种只要有少量样本就可以进行机器学习的相关方法的研究将对生物信息学领域的发展和应用起到至关重要的作用。
参考文献
- 庄福振, 中国科学院智能信息处理重点实验室, 庄福振,等. 迁移学习研究进展[J]. 软件学报, 2015, 26(1):26-39.
- 刘琦. 生物信息学研究的思考[R]. 中国计算机学会通讯, 2016, 12.10.
- 龙明盛. 迁移学习问题与方法研究[D]. 清华大学.
- PAN, Sinno Jialin; YANG, Qiang. A survey on transfer learning. IEEE Transactions on knowledge and data engineering, 2009, 22.10: 1345-1359.
- 何懿菡, 孙坤. 生物信息学研究进展[J]. 青海师范大学学报:自然科学版, 2011, 027(003):69-72.
- 张春霆. 生物信息学的现状与展望[J]. 世界科技研究与发展, 2000, 022(006):17-20.
- XU, Qian; YANG, Qiang. A survey of transfer and multitask learning in bioinformatics. Journal of Computing Science and Engineering, 2011, 5.3: 257-268.
- HANSON, Jack, et al. Identifying molecular recognition features in intrinsically disordered regions of proteins by transfer learning. Bioinformatics, 2020, 36.4: 1107-1113.
- PETEGROSSO, Raphael, et al. Transfer learning across ontologies for phenome–genome association prediction. Bioinformatics, 2017, 33.4: 529-536.
- MIGNONE, Paolo, et al. Exploiting transfer learning for the reconstruction of the human gene regulatory network. Bioinformatics, 2020, 36.5: 1553-1561.
- HURTADO, David Menéndez; UZIELA, Karolis; ELOFSSON, Arne. Deep transfer learning in the assessment of the quality of protein models. arXiv preprint arXiv:1804.06281, 2018.
- 赵屹, 谷瑞升, 杜生明. 生物信息学研究现状及发展趋势[J]. 医学信息学, 2012, 33(5):2-6.
| 版权声明本博客的文章除特别说明外均为原创,本人版权所有。欢迎转载,转载请注明作者及来源链接,谢谢。本文地址: https://blog.ailemon.net/2020/08/03/transfer-learning-in-bioinformatics/ All articles are under Attribution-NonCommercial-ShareAlike 4.0 |
关注“AI柠檬博客”微信公众号,及时获取你最需要的干货。

 WeChat Donate
WeChat Donate Alipay Donate
Alipay Donate
发表回复